
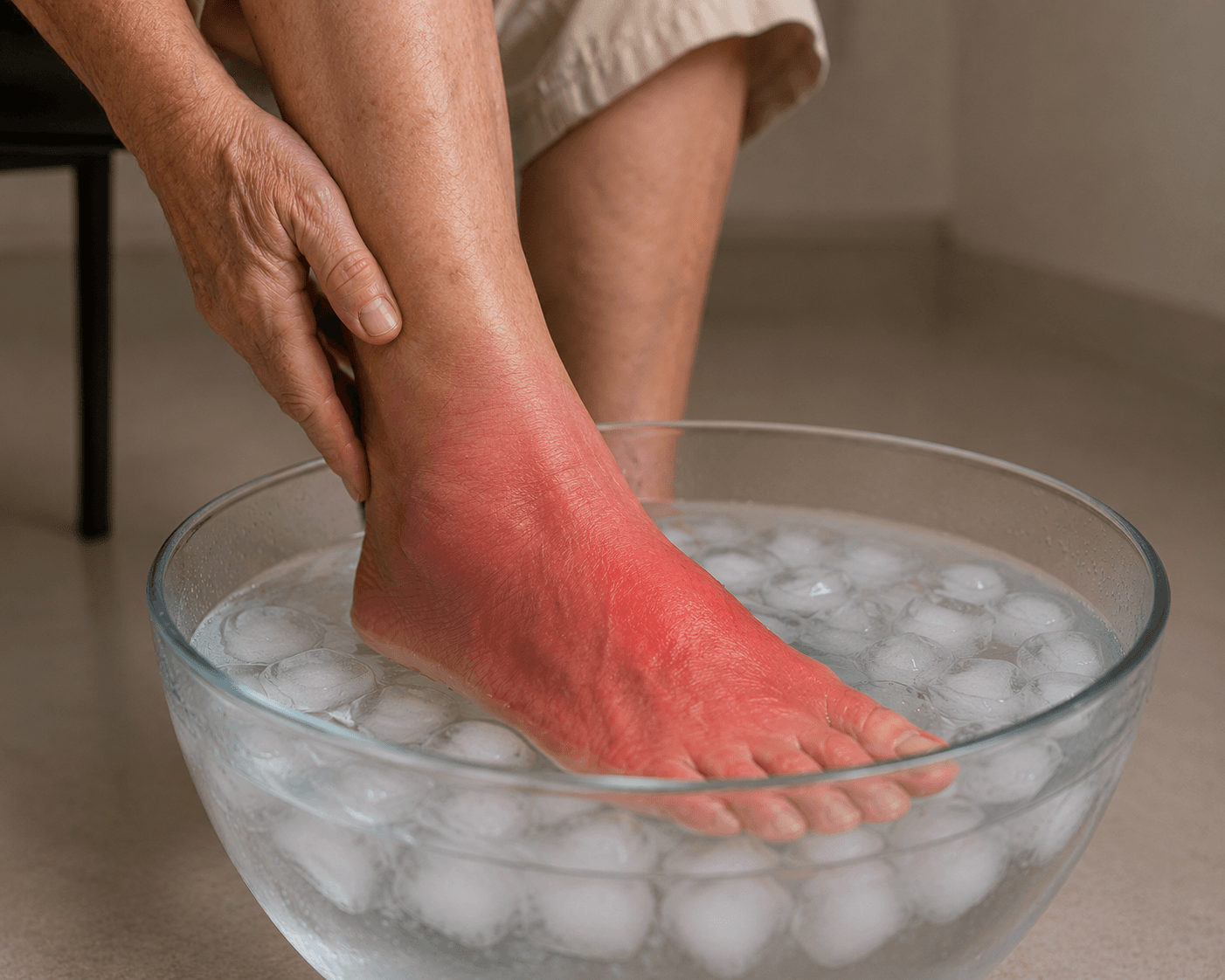
Mulher, 66 anos, com queimação e vermelhidão nos pés
Mulher de 66 anos apresenta episódios de queimação e vermelhidão nos pés, desencadeados por calor e aliviados pelo resfriamento. Qual será o diagnóstico final?
Autor: Carlos Antonio Moura
Data: 14 de maio de 2026
CASO CLÍNICO
Mulher de 66 anos, aposentada, hipertensa em uso de losartana 50 mg/dia, procura o ambulatório com queixa de episódios recorrentes de queimação intensa, vermelhidão e calor nos pés e nas pernas distalmente, com início há cerca de 8 meses. Os episódios duram entre 20 minutos e 2 horas, são desencadeados por calor ambiente, exercício físico leve e permanência de pé por tempo prolongado. A paciente relata que imergir os pés em água fria ou caminhar descalça em piso frio alivia a dor de forma significativa. Nega claudicação intermitente. Ao exame físico durante um episódio: extremidades distais com eritema intenso, temperatura aumentada ao toque, sem edema ou úlceras. Pulsos periféricos palpáveis e simétricos bilateralmente. Ausência de varizes significativas.
Questão 1. Qual síndrome clínica melhor descreve este quadro?
Comentário
A apresentação clínica descrita é clássica para eritromelalgia (alternativa B), condição caracterizada pela tríade de eritema, calor e dor em queimação nas extremidades distais – predominantemente os pés –, precipitada pela exposição ao calor e aliviada pelo resfriamento local. O próprio nome da síndrome reflete sua fenomenologia: erythros (vermelho), melos (membro) e algos (dor), do grego. Os episódios são tipicamente em crises, com duração variável de minutos a horas, e o comportamento de buscar superfícies frias ou imergir os pés em água gelada é um dado de anamnese altamente específico, frequentemente referido espontaneamente pelos pacientes.
A eritromelalgia pode ser classificada em primária ou secundária. A forma primária está associada, em parte dos casos, a mutações de ganho de função no gene SCN9A, que codifica o canal de sódio voltagem-dependente expresso em neurônios nociceptivos. A forma secundária, por sua vez, ocorre em associação a diversas condições sistêmicas, sendo as neoplasias mieloproliferativas – especialmente a policitemia vera e a trombocitemia essencial – as causas secundárias mais relevantes no adulto. O mecanismo fisiopatológico nesse contexto envolve ativação plaquetária com liberação de tromboxano A2 e oclusão microvascular transitória, gerando isquemia e dor nas extremidades. Por isso, toda eritromelalgia em adulto acima de 50 anos deve ser investigada com hemograma completo e pesquisa de neoplasia mieloproliferativa subjacente.
O fenômeno de Raynaud (alternativa A) representa um importante diagnóstico diferencial por também se manifestar como fenômeno neurovascular episódico nas extremidades. Pode ser primário (doença de Raynaud idiopática) ou secundário a conectivopatias, como esclerodermia e lúpus eritematoso sistêmico. Contudo, sua fisiopatologia e apresentação clínica são opostas à eritromelalgia: no Raynaud, o gatilho é o frio ou o estresse emocional, e o episódio cursa com vasoconstrição – manifestando-se como palidez seguida de cianose e, na fase de reaquecimento, rubor reativo (a tríade tricolor clássica). As extremidades estão frias ao toque, e o calor local alivia os sintomas – ao contrário do que ocorre na eritromelalgia, onde o calor é o gatilho e o frio é o alívio. A ausência de qualquer sequência de palidez-cianose-rubor e a piora com calor afastam o Raynaud neste caso.
A Doença arterial oclusiva periférica (DAOPC) (alternativa C) também entra no diferencial de dor em membros inferiores, mas seu padrão é completamente distinto. A isquemia arterial crônica cursa com claudicação intermitente – dor desencadeada pelo exercício, aliviada pelo repouso e predominantemente nas panturrilhas –, extremidades frias e pálidas, diminuição ou ausência de pulsos distais e, nos casos avançados, úlceras isquêmicas em áreas de pressão. Neste caso, os pulsos periféricos são palpáveis e simétricos, as extremidades estão quentes e hiperêmicas durante os episódios, e o repouso não é o elemento de alívio – o frio sim. Esses dados são incompatíveis com isquemia arterial macrovascular.
A neuropatia de fibras finas (alternativa D) é o diferencial que mais se aproxima clinicamente da eritromelalgia, pois ambas cursam com dor em queimação nos pés. As neuropatias de pequenas fibras acometem fibras C e Aδ não mielinizadas, responsáveis pela nocicepção e pela regulação autonômica vascular, e podem causar disestesias, alodinia e queimação nos pés, especialmente à noite. No entanto, a neuropatia de pequenas fibras tipicamente não se acompanha dos sinais vasomotores objetivos da eritromelalgia – o eritema intenso, o aumento mensurável de temperatura e o alívio consistente com resfriamento. Quando presentes juntos, a distinção requer biópsia de pele – e não de nervo – para quantificação da densidade de fibras nervosas intraepidérmicas e testes de função autonômica.
CONTINUAÇÃO DO CASO
Durante a anamnese detalhada, a paciente refere também prurido intenso após banho quente, presente há aproximadamente 6 meses, sem lesões dermatológicas visíveis. Relata sensação de plenitude epigástrica ocasional. Nega icterícia, perda de peso significativa ou sudorese noturna. Ao exame físico: peso 72 kg, altura 1,61 m, PA 148/90 mmHg. Sem icterícia, sem linfonodomegalias. Fígado palpável a 1 cm do rebordo costal direito com hepatimetria normal; baço não palpável. Sem sinais de hepatopatia crônica.
Questão 2. O prurido aquagênico (após contato com água) associado à eritromelalgia nesta paciente sugere qual diagnóstico?
Comentário
A associação de eritromelalgia com prurido desencadeado especificamente pelo contato com água – o chamado prurido aquagênico – é altamente sugestiva de PV (alternativa C). Trata-se de duas das manifestações mais características dessa neoplasia mieloproliferativa, e sua concomitância deve levar à suspeita imediata, independentemente dos valores de hemoglobina. O prurido aquagênico ocorre em 40 a 70% dos pacientes com PV e resulta da degranulação de mastócitos e basófilos – células derivadas do mesme clone neoplásico – com liberação de histamina, leucotrienos e prostaglandinas em resposta ao contato com água. É um mecanismo independente de IgE, portanto distinto das reações alérgicas convencionais. A intensidade do prurido pode ser incapacitante, levando pacientes a evitar banhos e impactando severamente a qualidade de vida.
A dermatite atópica do adulto (alternativa A) é uma dermatose inflamatória crônica mediada por hipersensibilidade do tipo Th2, caracterizada por prurido intenso associado a lesões eczematosas – eritema, vesiculação, descamação e liquenificação – distribuídas em padrão típico (face, pescoço, fossas antecubitais e poplíteas). A ausência completa de lesões cutâneas visíveis é um elemento fundamental neste caso e afasta a dermatite atópica, que invariavelmente cursa com alterações dermatológicas objetivas. Além disso, a atopia costuma ter início ainda na infância ou adolescência, e a história prévia de rinite alérgica, asma ou eczema seria esperada.
A colestase intra-hepática (alternativa B) é uma causa reconhecida de prurido – chamado prurido colestático –, decorrente do acúmulo de ácidos biliares e outros compostos na pele, com ativação de fibras nervosas pruritoceptivas. Contudo, o prurido colestático quase sempre se acompanha de icterícia, acolia, colúria e alterações laboratoriais características (hiperbilirrubinemia, elevação de fosfatase alcalina e gama-GT). Além disso, o prurido colestático não tem relação específica com água – pode piorar ao anoitecer e com o calor, mas não é desencadeado exclusivamente pelo banho. A ausência de icterícia e a especificidade aquagênica do prurido desta paciente afastam a colestase como causa principal.
A urticária colinérgica (alternativa D) é uma forma de urticária física desencadeada pelo aumento da temperatura corporal – seja pelo exercício físico, banho quente ou estresse emocional. Manifesta-se com pápulas urticariformes pequenas (1 a 5 mm), eritematosas, rodeadas de halo, que surgem minutos após o gatilho e regridem em 30 a 60 minutos. Embora possa ser precipitada pelo banho quente, há uma distinção importante: na urticária colinérgica, o agente desencadeante é o calor e a sudorese, e as lesões cutâneas são objetivamente visíveis; no prurido aquagênico da PV, o simples contato com água desencadeia o prurido sem a presença de lesões urticariformes. A ausência de pápulas visíveis nesta paciente afasta a urticária colinérgica.
CONTINUAÇÃO DO CASO
O médico solicita hemograma completo e outros exames laboratoriais. Os resultados revelam: Hemoglobina 15,4 g/dL (VR feminino: 12,0–16,0 g/dL) – discreta elevação no limite superior; Hematócrito 49% (VR: 36–46%); VCM 78 fL (VR: 80–100 fL) – microcitose leve; HCM 26 pg (VR: 27–33 pg); Leucócitos 12.800/mm³ (VR: 4.000–10.000), com diferencial normal; Plaquetas 520.000/mm³ (VR: 150.000–400.000); Ferritina 8 ng/mL (VR: 12–150); Ferro sérico baixo; TIBC aumentado. Ultrassonografia abdominal: baço de dimensões normais (9,8 cm), sem esplenomegalia. Pesquisa de sangue oculto nas fezes: positiva em duas amostras.
Questão 3. Como interpretar este quadro no contexto diagnóstico?
Comentário
Este é um dos cenários mais desafiadores e didaticamente mais ricos na investigação da policitemia vera: a possibilidade de a eritrocitose estar mascarada por deficiência de ferro concomitante (alternativa B). Na PV, a medula óssea produz eritrócitos em quantidade aumentada, sob estímulo da clona JAK2-mutante. Se, simultaneamente, há privação de ferro por sangramento crônico, a hemoglobinização dos eritrócitos torna-se limitada, resultando em células microcíticas e hipocrômicas. O organismo mantém a eritropoiese aumentada, mas os eritrócitos produzidos carregam menos hemoglobina. O efeito líquido é uma hemoglobina que pode parecer "normal" ou apenas discretamente elevada, enquanto a clona neoplásica prolifera ativamente. Neste caso, a hemoglobina associada microcitose (VCM 78 fL), ferritina de 8 ng/mL e sangue oculto positivo configura exatamente essa situação. Se não houvesse o sangramento, a hemoglobina poderia estar significativamente mais elevada.
A afirmação de que a hemoglobina normal exclui a PV (alternativa A) é incorreta e potencialmente prejudicial ao paciente. Os critérios diagnósticos da OMS (2022) para PV contemplam valores de hemoglobina ≥16,5 g/dL em homens e ≥16,0 g/dL em mulheres como critério de eritrocitose – mas reconhecem explicitamente que esses valores podem estar abaixo do limiar quando há deficiência de ferro, sangramento ou hemodiluição. Ainda existe a possibilidade de anemia em pacientes com PV que cursam com a transformação para mielofribrose.
Nesses casos, o hematócrito (≥49% em homens, ≥48% em mulheres) e, preferencialmente, a dosagem direta da massa eritrocitária por cromo radioativo (Cr51) são mais informativos. A mensagem clínica fundamental é: hemoglobina normal não exclui PV quando há suspeita clínica consistente e fatores que possam mascarar a eritrocitose.
Atribuir o quadro exclusivamente à anemia ferropriva por sangramento digestivo (alternativa C) seria um erro de raciocínio clínico por insuficiência explicativa. A deficiência de ferro isolada não justifica leucocitose (12.800/mm³), ainda que explique trombocitose. A eritromelalgia recorrente há 8 meses e prurido aquagênico há 6 meses não são sintomas usuais de neoplasia gastrointestinal. Cada um desses elementos, isoladamente, poderia ter outra explicação, mas a concomitância de todos eles em uma única paciente aponta para uma causa sistêmica comum – a neoplasia mieloproliferativa. Ressalte-se que o sangramento digestivo oculto pode ser uma complicação da própria PV – dada a disfunção plaquetária qualitativa que acompanha a trombocitose e consequente consumo do fator de von Willebrand.
A hipótese de trombocitemia essencial (alternativa D) merece atenção, pois integra legitimamente o diagnóstico diferencial. A TE também é uma neoplasia mieloproliferativa JAK2-positiva (em cerca de 50 a 60% dos casos), pode causar eritromelalgia, trombocitose e, menos frequentemente, prurido aquagênico. No entanto, a TE caracteriza-se mais pelo predomínio isolado da linhagem megacariocítica, sem comprometimento expressivo da série vermelha. A presença de hematócrito de 49% – acima do limiar diagnóstico para PV em mulheres – e a microcitose com deficiência de ferro, sugerindo eritrocitose mascarada, inclinam a balança para PV. A distinção definitiva entre PV e TE requer a integração de mutação JAK2, dosagem de eritropoietina sérica e biópsia de medula óssea, que na PV mostra panmielose com proliferação das três séries, enquanto na TE predomina a hiperplasia megacariocítica com arquitetura medular preservada.
CONTINUAÇÃO DO CASO
Diante da suspeita de policitemia vera, o médico solicita exames: eritropoietina sérica (EPO): 2,1 mUI/mL (VR: 4,3–29,0) – suprimida; Mutação JAK2 V617F: positiva (carga alélica 42%); Biópsia de medula óssea: hipercelularidade acentuada com proliferação das três séries (panmielose), megacariócitos pleomórficos e agrupados – compatível com policitemia vera. Critérios diagnósticos OMS 2022 preenchidos (3 critérios maiores: hematócrito ≥49%, biópsia compatível, JAK2+). O diagnóstico de Policitemia Vera é estabelecido. Risco cardiovascular estratificado como alto risco (idade >60 anos). A paciente pergunta sobre o prognóstico e os riscos. O médico explica que PV não tratada tem risco significativo de trombose (acidente vascular encefálico, tromboembolismo pulmonar, trombose de veia porta) e que o objetivo do tratamento é reduzir esse risco.
A paciente inicia flebotomias quinzenais, hidroxiureia 500 mg/dia e AAS 100 mg/dia. Após 3 semanas, relata melhora expressiva dos episódios de eritromelalgia. O prurido aquagênico também reduziu significativamente. O hematócrito cai para 46% após duas flebotomias. O hemograma de controle após 8 semanas mostra: Hb 14,2 g/dL, Ht 43%, leucócitos 8.400/mm³, plaquetas 390.000/mm³. A deficiência de ferro é tratada com suplementação oral após controle endoscópico do sangramento. A colonoscopia revela angiodisplasia de cólon direito, tratada com coagulação a plasma de argônio.
CONCLUSÃO
O caso apresentado ilustra uma forma de apresentação incomum e didaticamente valiosa da policitemia vera, na qual as manifestações iniciais – eritromelalgia recorrente e prurido aquagênico – precederam em meses o reconhecimento do diagnóstico hematológico subjacente. A “aparente normalidade” da hemoglobina, explicada pela concomitância de deficiência de ferro secundária a sangramento digestivo crônico por angiodisplasia, representa uma das principais armadilhas diagnósticas da PV e reforça o conceito de que a ausência de eritrocitose franca jamais deve afastar a suspeita diagnóstica quando o contexto clínico é sugestivo. A investigação com mutação JAK2 V617F e eritropoietina sérica deve ser iniciada precocemente diante de eritromelalgia inexplicada, prurido aquagênico, leucocitose e trombocitose, mesmo na ausência de hemoglobina expressivamente elevada. O tratamento com a tríade hidroxiureia, AAS e flebotomia resultou em controle rápido e satisfatório dos sintomas, com resolução da eritromelalgia e redução do prurido aquagênico, destacando o papel central do AAS na fisiopatologia microvascular da doença.
Um mnemônico útil para lembrar de como a PV pode se apresentar é o P.V.E.R.A (Policitemia; fenômeno Vasomotor; Esplenomegalia; Recorrência de trombos; AVCs)
REFERÊNCIAS
1. Tremblay D, Kremyanskaya M, Mascarenhas J, Hoffman R. Diagnosis and Treatment of Polycythemia Vera: A Review. JAMA. 2025;333(2):153–160. doi:10.1001/jama.2024.20377
2. Bewersdorf JP, How J, Masarova L, Bose P, Pemmaraju N, Mascarenhas J, Rampal RK. Moving toward disease modification in polycythemia vera. Blood. 2023 Nov 30;142(22):1859-1870. doi: 10.1182/blood.2023021503.
3. Bennett C, Jackson VE, Pettikiriarachchi A, Hayman T, Schaeper U, Moir-Meyer G, Fielding K, Ataide R, Clucas D, Baldi A, Garnham AL, Li-Wai-Suen CSN, Loughran SJ, Baxter EJ, Green AR, Alexander WS, Bahlo M, Burbury K, Ng AP, Pasricha SR. Iron homeostasis governs erythroid phenotype in polycythemia vera. Blood. 2023 Jun 29;141(26):3199-3214.
4. DAVIS, M. D. P.; SANDRONI, P. Erythromelalgia: vascular disease, neuropathy, or both? Archives of Dermatology, v. 139, p. 1337–1343, 2003.
Comentários (0)
Nenhum comentário ainda. Seja o primeiro!
Faça login para deixar um comentário.
Entrar